Troubleshooting¶
The most common error comes from unstable Internet connection, especially during downloading genomes (first run). Simply close and run FREEDA again.
- In case your issue is not covered here please send (damiandudka0@gmail.com):
Fatal errors (action needed)¶
...FATAL ERROR... : Something went wrong (see below). Send screen shot to : Damian Dudka -> damiandudka0@gmail.com
REASON: Follows most unknown errors causing FREEDA to crash.
SOLUTION: Please attach a screen shot and "FREEDA-current-date.log" and (if present) "PAML-current-date.log". They will be either in "Raw_data" folder or in the main folder indicated within the GUI ("Set directory").
...FATAL ERROR... : PyMOL not found in the PATH, the Applications folder, or the working directory.
REASON: This is most likely a iOS user error installing PyMOL outside of the Applications folder.
SOLUTION: Move the PyMOL .app to Applications.
...FATAL ERROR... : Partial or empty genome file : GENOME.fasta -> likely Internet connection was disrupted -> please run FREEDA again...
REASON: The most common error at first run - almost always caused by unstable Internet connection or closing the app when downloading the genomes at first run.
SOLUTION: Simply re-run the app.
...FATAL ERROR... : Genome : GENOME blast databases failed to build (likely interrupted) -> rerun the pipeline...
REASON: Most likely the app crashed or was closed while making local BLAST databases.
SOLUTION: Try re-running the app.
...FATAL ERROR... : Genome : GENOME blast databases were built partially (likely interrupted) -> rerun the pipeline...
REASON: Similar to the previous error. FREEDA is expecting a certain size of the generated databases. Partial files may be present when the run was interrupted.
SOLUTION: Try re-running the app.
...FATAL ERROR... : Repetitive coding sequence detected in GENE (min 80bp repeat) -> cannot reliably analyze this GENE
REASON: FREEDA's ability to find orthologous exons relies on accurate alignment to the reference coding sequence. Repetitive sequences are notoriously difficult to align, therefore FREEDA will not try to analyze genes with more than 80bp-long repeats in coding sequence (e.g. Tacc3 in rodents).
SOLUTION: Unfortunately this gene cannot be analyzed reliably. If you still want to try I can run the analysis on my end and let you know if its reliable: damiandudka0@gmail.com
...FATAL ERROR... : GENE is transposable element instead of a protein coding gene -> check gene name for SPECIES here: ensembl.org -> correct gene name and click Analyze again...
REASON: Provided gene name does not encode a protein (but its a trasposable element in this case).
SOLUTION: Check out the link FREEDA gives you and ensure you know the name of the protein coding gene you are interested in. ALWAYS use the release FREEDA gives you the link to.
...FATAL ERROR... : No reliable coding sequence annotation detected for GENE
REASON: This is usually caused by unreliable annotation of the gene of interest - mostly concerning dog and chicken assemblies.
SOLUTION: Verify your gene annotation that FREEDA used under the link provided
...FATAL ERROR... : Gene names [GENE] do not exist in reference assembly -> check gene name for SPECIES here: ensembl.org -> correct gene name and click Analyze again...
REASON: This is usually caused by unreliable annotation of the gene of interest - mostly concerning dog and chicken assemblies.
SOLUTION: Make sure there is a known coding sequence for this gene (e.g. visit Ensembl database - https://useast.ensembl.org/index.html
...FATAL ERROR... : Input data generation FAILED for GENE - please remove GENE from analysis -> exiting the pipeline now...
REASON: Unreliable input generation - usually following another FATAL_ERROR (e.g. no coding transcript found)
SOLUTION: Make this is not a pseudogene (e.g. visit Ensembl database - https://useast.ensembl.org/index.html
...FATAL ERROR... : >= 5 species show no matches above blast threshold: 60% Gene: GENE -> please run FREEDA again with Exclude species option (copy paste this: Pt Gg)
REASON: FREEDA uses a default 60% (vertebrates) or 30% (flies) treshold for identity when blasting coding sequences. In case of very divergent genes that threshold might not be met. If you used "Common domains expected" advanced option that increases the threshold to 80% (vertebrates) or 60% (flies) increasing the risk of no matches found for a given species. It might also be due to a gene loss in some species.
SOLUTION: Try the "Exclude species" option using two-letter (or three-letter for flies) code for each genome (e.g., type Pt Gg for Pan troglodytes and Gorilla gorilla; or Dsi Der for Drosophila simulans and Drosophila erecta). You can also try to indicate a subgroup (hominoidea, catarrhini, caniformia, melanogaster) to avoid blasting against more divergent species.
...FATAL ERROR... : Too few genomes were selected (< 6)
REASON: Similar to the previous error. FREEDA expects coding sequences from at least 6 species (genomic assemblies) to be able to run the analysis. This is most likely to happen if you select melanogaster as subgroup (maximum 9 species available).
SOLUTION: Make sure not to select "Common domains expected" and do not select melanogaster subgroup (allowing search in more divergent flies too).
Warnings (no action needed)¶
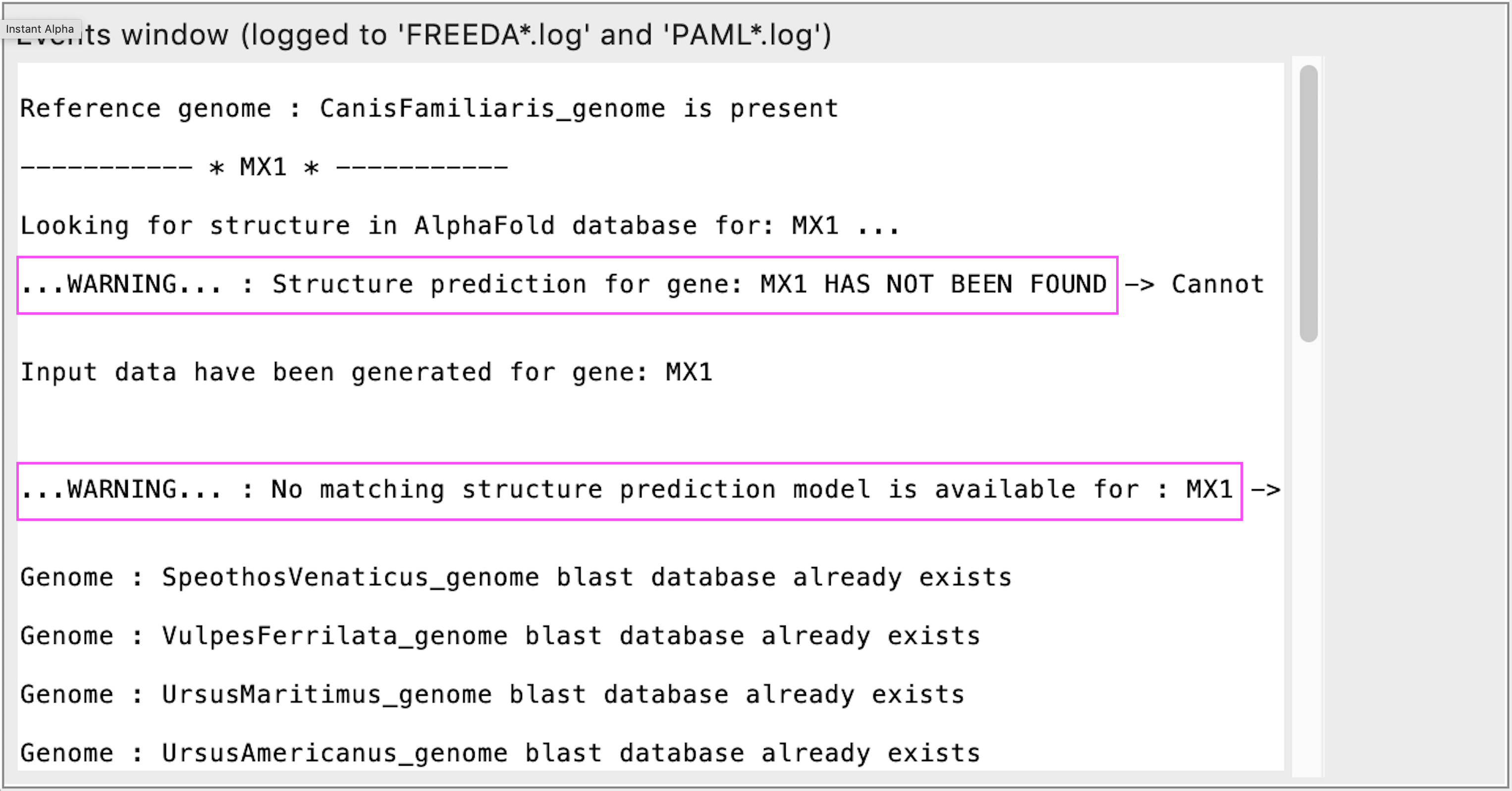
AlphaFold structure not found or not matching Ensembl input collected
REASON: FREEDA will still run the analysis but without mapping residues onto structure
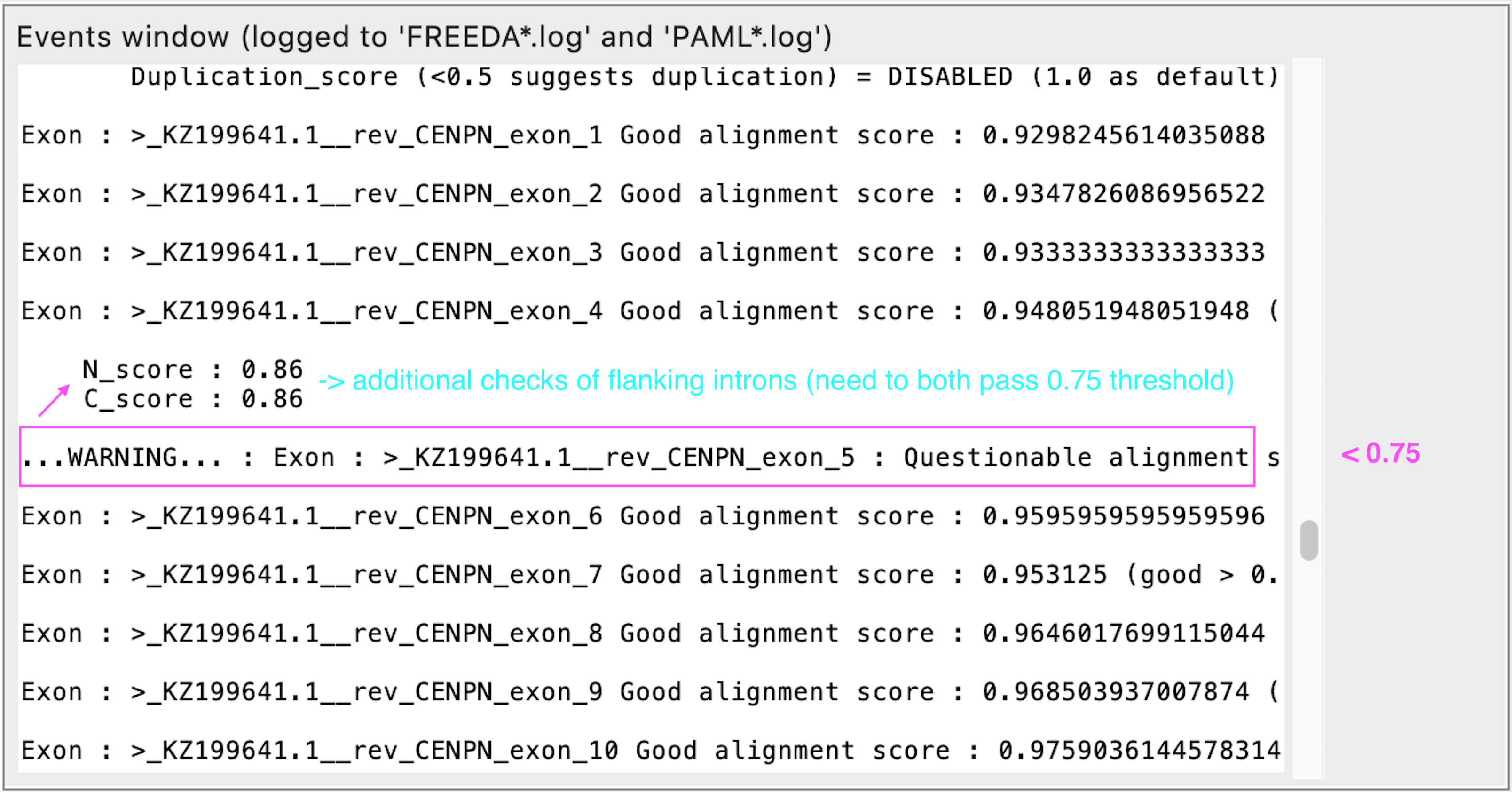
Questionable alignment of a single exon
REASON: FREEDA performs additional checks (blue) for a divergent exon and accepts or rejects it
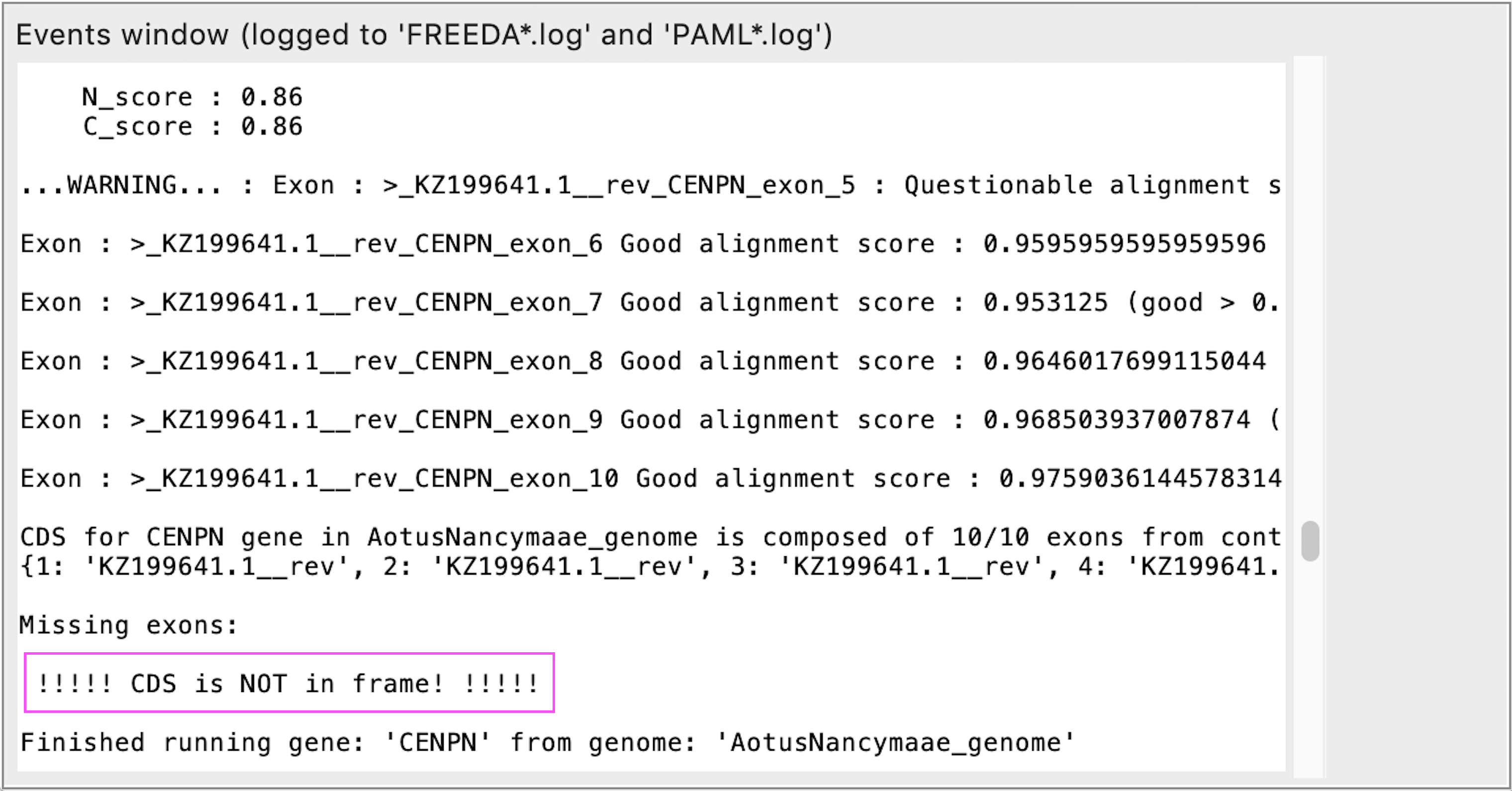
Coding sequence is not in frame
REASON: Either some exons are missing (not the case in example below) or single indels are present (e.g. sequencing errors). FREEDA may either remove this sequence from analysis or remove the indels to force conserved alignment.
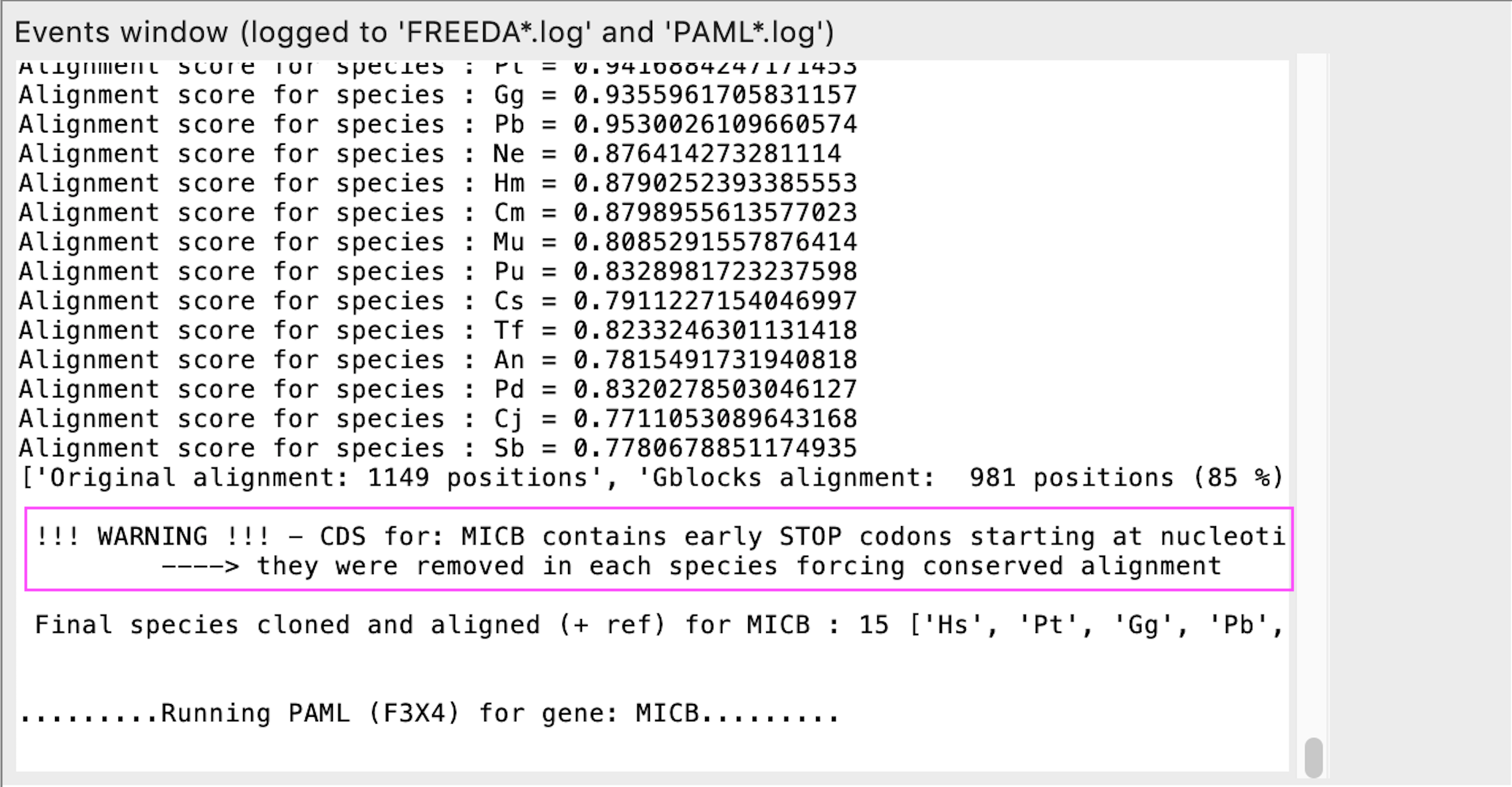
Early STOP codon detected in final coding sequence
REASON: Coding sequences in the final alignment should not have any STOP codons anymore, except those introduced by imperfect alignment. This is rare and usually occurs in highly divergent genes. FREEDA removes the STOP codon forcing a conserved alignment.
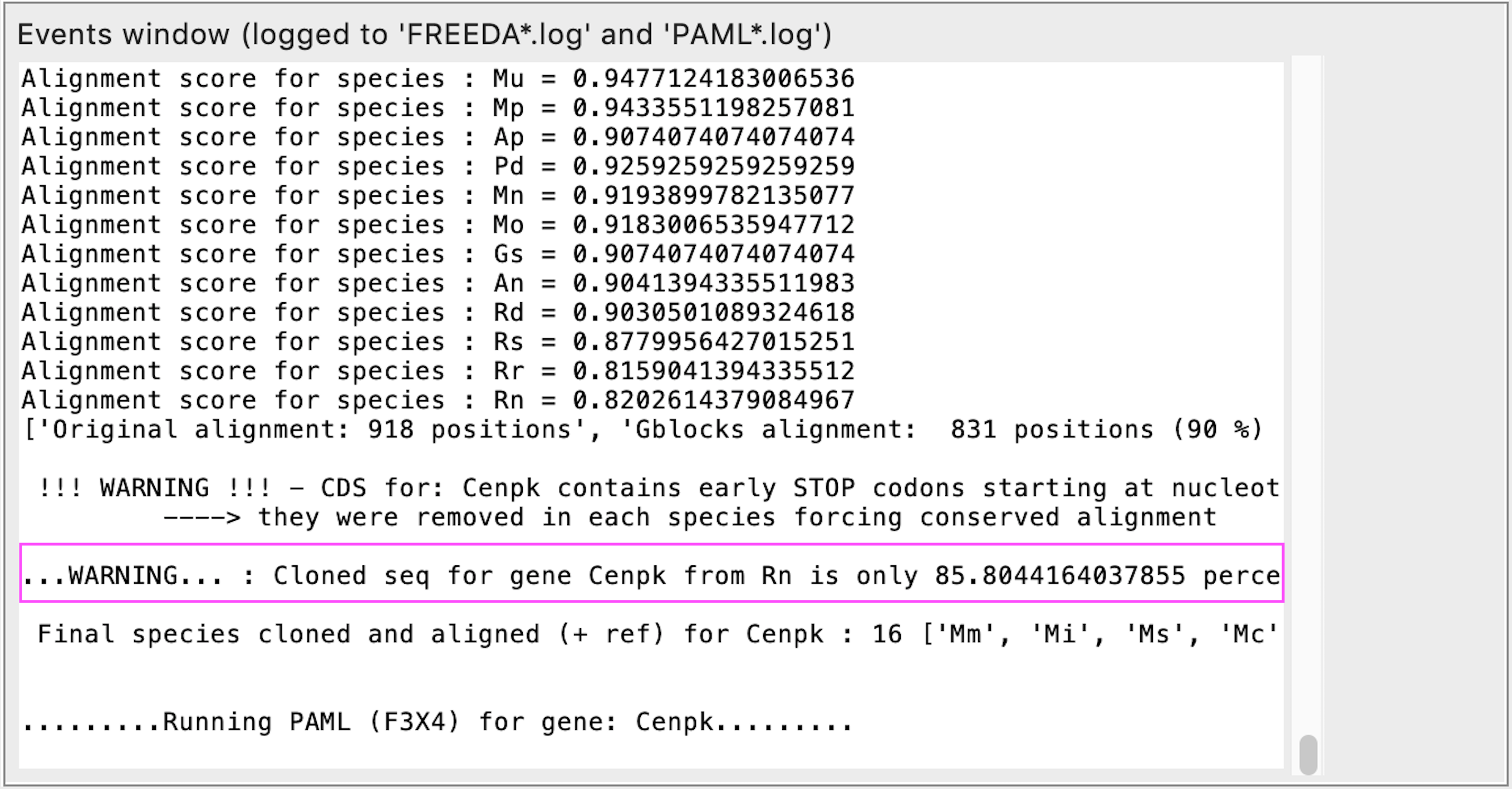
Failed check comparing cloned sequence to annotated one for most distant species
REASON: This is a sanity check. Usually <95% identity suggests that alternative exons are used (check supported only for rodents and carnivores).
Problems with VirtualBox¶
64-bit Operating Systems Won't Show Up¶
Your Windows system may already be using another Virtual Machine system called HyperV.
Other problems¶
PyMOL desktop file may not work¶
Ubuntu 22.04 LTS version does not seem to support giving launching permissions to Desktop files but that might change with future updates. For now just double-click on .pse files to open structures in PyMOL on Linux.
PyMOL Tar Unpacking "Cannot Create Symlink"¶
Your data folder may be in a system partition format that does not allow symbolic links. As a work around, you can install PyMOL yourself from a package manager, software application, or the open source PyMOL repository: https://github.com/schrodinger/pymol-open-source <https://github.com/schrodinger/pymol-open-source>.